
The COVID-19 crisis has affected multiple industry sectors and disrupted economic processes. Because of the increased burden that the pandemic has placed on the medical device industry, the application of the Medical Device Regulation (“MDR”) was postponed by one year. It will now apply from May 26 2021 (instead of 2020). Since the MDR also regulates software which can qualify as a medical device, this extended timeline provides developers of mobile medical applications the opportunity to (re-)evaluate the classification of their app and to implement any MDR required changes. The following article takes a closer look at the requirements that the MDR imposes on health apps or medical apps and compares these with the requirements for medical apps under the US regulatory regime.
1. Development stage – Does the app need regulatory approval?
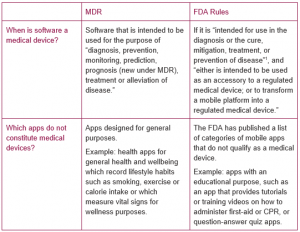
A health app requires regulatory approval either in the EU under the MDR or in the USA under FDA (Food & Drug Administration) rules when the app qualifies as a “medical device”.
2. If the app meets the criteria of “medical device”: Classification of the app to determine what regulatory approval is necessary, and whether self-certification is possible
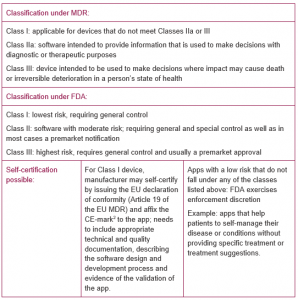
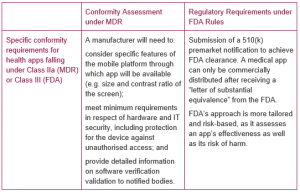
3. Regulatory approval or clearance
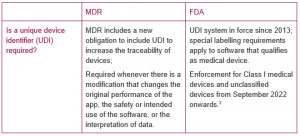
4. UDI (Unique Device Identifier)
5. Post-Market Surveillance after Launching the App
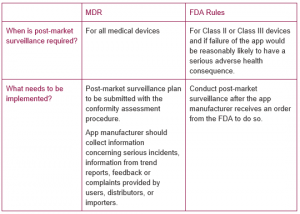
Conclusion
Any app developer that wants to market an app relating to wellbeing, health or medical treatments either in Europe or the US needs to be aware of the impact that the MDR or the FDA Rules have on their business. Developers intending to launch in Europe should ensure that they have a clear strategy underway to achieve compliance in time for May 2021.
for any question, please contact
Simon Marks or Dr. Laura Jelinek
[1] FDA guidance document, published in September 2019: Policy for Device Software Functions and Mobile Medical Applications – Guidance for Industry and Food and Drug Administration Staff.
[2] With the CE mark a manufacturer expresses conformity with the European legislation, specifically with European directives and European regulations.
[3] FDA guidance document, published on July 1, 2020: Unique Device Identification: Policy Regarding Compliance Dates or Class I and Unclassified Devices and Certain Devices Requiring Direct Marking